Snakemake (https://snakemake.readthedocs.io/en/stable/index.html)
Exonerate (https://www.ebi.ac.uk/about/vertebrate-genomics/software/exonerate) (v.2.4.0)
Perl (https://www.perl.org/get.html) (v5.30.0)
AWK
Java
Parallel (https://manpages.ubuntu.com/manpages/impish/man1/parallel.1.html)
Perl Modules
Getopt::Long
Getopt::Std
Parallel::ForkManager
Make sure you have all dependencies installed. You also need to download and have in your path all the "bin" scripts.
To avoid errors with Java, you also need to create a variable with the absolute path of "readseq.jar" which is in the bin folder:
export CLASSPATH="/full/path/to/bin/readseq.jar"
You can check Snakemake on their site for more details of this.
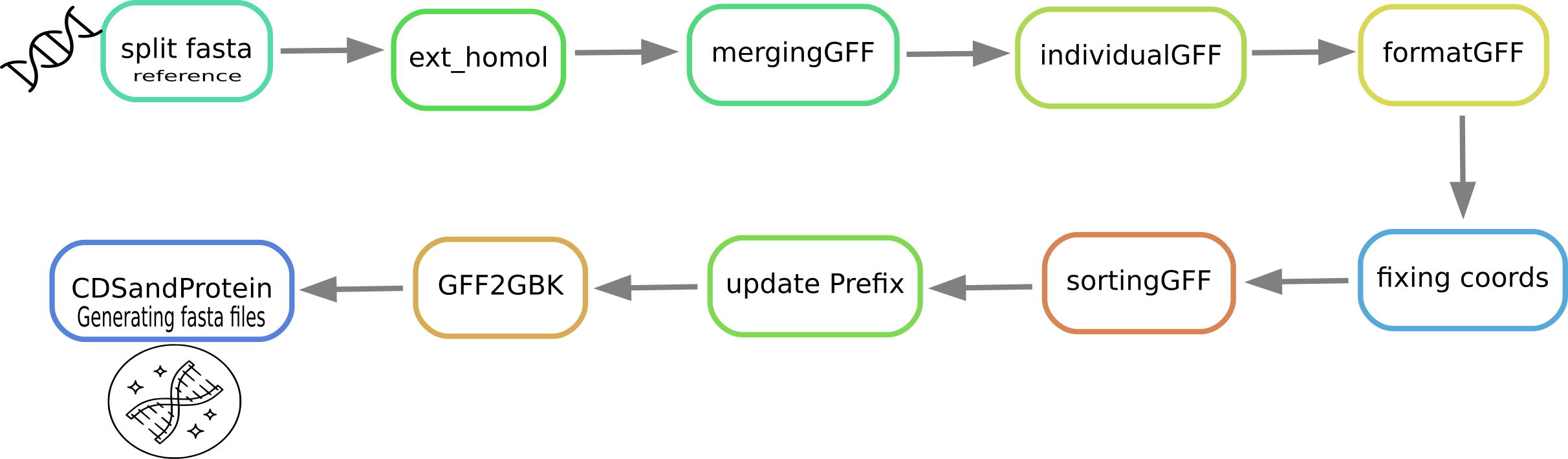
For genome.fasta and protein.faa file name run:
snakemake --cores -s /path/of/Snakefile
If genome or protein fastas files have other names, then run:
snakemake --cores <core_numbers> --config PROTREF="current_protein_fasta_filename" GENOME="current_genome_fasta_filename" -s path/of/Snakefile_PATT
snakemake --cores <core_numbers> --rerun-incomplete --config PROTREF="protein.faa" GENOME="genome.fasta" PREFIX="prefix_outputfilename" NEWPREFIX="prefix_newgenenames_" -s path/of/Snakefile_PATT
About variables that PATT optionally needs:
GENOME= "genome.fasta" # Fasta file of genome that we want to annotate. Default: "genome.fasta"
PROTREF= "protein.faa" # Fasta file of the reference proteins that we want to transfer or annotate in our genome. Default: "protein.faa"
PREFIX= "prefix" # Output file prefix. Default: "mySpecies"
NEWPREFIX= "prefix_" # Prefix name we want for the proteins/transcripts in the our genome. We suggest ending in "" for aesthetics. Default: "{PREFIX}"
OLDPREFIX= "prefix" # Prefix that the proteins have in the faa to be transferred. Perl regular expressions are accepted ex: "^\S+gene[^_|\s]+". PATT generates new names of the transferred proteins keeping all(default) or a part of the original annotated protein identifier. Default: "=gene". ex: if the names of the proteins to be transferred have this form "tsol_\d+", my variable can be OLDPREFIX= "=genetsol_" and the new names will be "mySpecies_\d+"
The output of PATT produces 4 files:
Annotation file in GFF format of the transferred proteins.
Annotation file in GenBank format of the transferred proteins.
Fasta file of all coding sequences (CDs).
Fasta file of the peptide sequences.
Estrada, K. (2023). PATT (Proteome Annotation Transfer Tool) (Version 1) [Computer software]. https://doi.org/10.5281/zenodo.7958134
PATT wouldn't be the same without my fellow researchers at the UUSMB (Unidad Universitaria de Secuenciación Masiva y Bioinformática) Jerome Verleyen and Alejandro Sanchez, who helped me with ideas and challenges during PATT's development.
PATT uses Snakemake for pipeline development, Exonerate to perform alignments, Readseq for handling file formats, Mario Stanke script "gff2gbSmallDNA.pl" and many lines of code and scripts from my dear friend and god-level programmer, Alejandro Garciarrubio, I am grateful for his help and guidance.
Karel Estrada
Twitter: @kjestradag